

Diamond-Blackfan Anemia (DBA) is a rare, inherited bone marrow (BM) failure syndrome characterized by pure red cell aplasia generally presenting within the first year of life. The majority of cases are due to heterozygous mutations in ribosomal genes, either autosomal dominant inherited or sporadicde novoevents. Despite the recognition of the importance of ribosomal biology, the underlying mechanisms are still poorly understood. The only curative treatment is hematopoietic stem cell transplantation, otherwise steroids or transfusions will cause significant long-term morbidity. There is an unmet need for new therapies.
We describe the unique clinical course and molecular analyses of a 32-year-old Hispanic woman, provide insights into disease pathophysiology as well as hope for an effective new treatment. She was diagnosed with DBA at the age of one month with isolated severe normochromic anemia. She failed various therapies, including corticosteroids, danazol, growth factors, and additional immunosuppression and developed significant iron overload despite desferal chelation. She was enrolled in a phase II clinical trial investigating the safety and efficiency of the thrombopoietin receptor agonist eltrombopag (EPAG) in patients with moderate aplastic anemia or unilineage cytopenias (NCT01328587). EPAG was administered in an escalating manner reaching 300mg daily. She achieved transfusion-independence by 16 weeks. This dose was continued for an additional 6 months and discontinued for protocol-defined robust response, ie Hb >10g/dL. After discontinuation, her hemoglobin steadily declined over a period of 8 months. EPAG was reinitiated, her Hb again rapidly improved and the EPAG dose was slowly tapered. Seven years since enrollment, the patient has remained on drug, currently at a dose of 25mg daily with normal blood cell counts and no EPAG related toxicity. BM cytogenetics and morphology have remained normal throughout follow up, as has a myeloid somatic mutation panel.
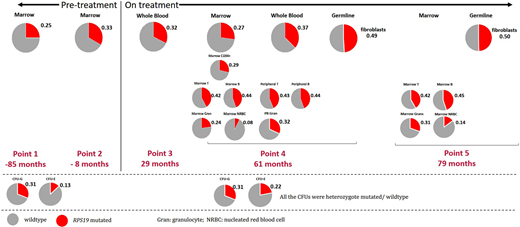
Next generation sequencing has revealed a novel, pathogenic mutation in intron 4(c.356+3>C) of theRPS19gene affecting a splice donor site previously implicated in DBA. Testing of both parents was negative, documenting this to be ade novomutation. With digital droplet PCR (ddPCR), we have monitored the variant allele frequency (VAF) in fibroblasts (germline) and different hematopoietic populations (Figure 1). Fibroblasts showed a VAF 50% (heterozygous), while her whole BM and whole blood show a lower VAF between 25% and 37%, both before and on EPAG treatment. Across lineages were analyzed when she was on EPAG, lymphocytes showed the highest VAF (42% to 45%), followed by HSPCs and granulocytes at 24% to 32%. Of note, the erythrocytes progenitors (CD45-CD71+) had the lowest VAF of 8% to 14%. Sequencing of myeloid and erythroid colony forming units (CFU) with both pre and on EPAG BM samples confirmed only heterozygous mutations or wild-type (WT) CFUs in percentages comparable to their respective VAF detected by bulk ddPCR. These results suggest the emergence of WT clones within the BM either through mosaicism or a reversion event. The discrepancy in VAF between peripheral and BM samples is consistent with a proliferative advantage for WT clones, especially in the erythroid and myeloid lineages. Yet despite the presence of WT HSPCs, profound transfusion-dependent anemia persisted in this patient long-term. An imbalance between globin chain and heme production has been documented to result from ribosomal dysfunction in DBA, leading to increased iron-mediated cellular stress and death of erythroid progenitors. We hypothesize the residual mutant cells suppress the growth of WT cells, given the close proximity of large numbers of erythroid cells aggregated by macrophages in erythroid islands, potentially via passage of free iron. These observations have implications for the development of gene addition or gene correction therapies for DBA, suggesting that correcting a fraction of HSPCs will not revert the phenotype.
Given the potent intracellular chelation activity of EPAG, and the lack of the EPAG target MPL receptor on erythroid progenitors, we hypothesize that iron chelation is a potential mechanism for improvement of red blood cell production by EPAG in DBA. The results in our patient have encouraged the opening of a prospective clinical trial (NCT04269889) to further explore the potential of EPAG for DBA patients.
Young:Pharos I&BT Co., Ltd.:Research Funding.Pakbaz:Amgen:Membership on an entity's Board of Directors or advisory committees;Dova:Membership on an entity's Board of Directors or advisory committees;Novartis:Membership on an entity's Board of Directors or advisory committees, Speakers Bureau;Terumo BCT:Membership on an entity's Board of Directors or advisory committees, Speakers Bureau;Global Blood Therapeutics:Speakers Bureau.Dunbar:Novartis:Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal